Cu-Catalyzed Direct Amination of Cyclic Amides via C–OH Bond Activation Using DMF
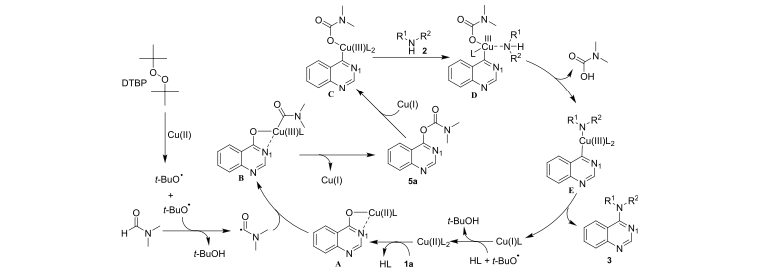
Herein, we describe a Cu-catalyzed approach to directly accessing aromatic heterocyclic amines from cyclic amides. The most-reported methods for cyclic amide conversions to aromatic heterocyclic amines use an activating group, such as a halogen atom or a trifluoromethane sulfonyl group. However, subsequent elimination of activating groups during the amination process results in significant waste. This copper-catalyzed direct amination of cyclic amides in DMF forms aromatic heterocyclic amines with environmental friendliness and readily available reagents. A plausible radical mechanism has been proposed for the reaction. Meanwhile, the coordinating effect of the N1 atom is key to the success of this reaction, which provides assistance to the copper ions for the activation and amination of C–O bonds.
查看文献